 |

Витяев Е.Е.
Институт математики СО РАН, Новосибирск
Орлов Ю.Л., Вишневский О.В., Поздняков М.А.,
Беленок А.С.,
Подколодный Н.Л.,
Колчанов Н.А.
Институт цитологии и генетики СО РАН, Новосибирск
Ковалерчук Б.К.
Central Washington University, Ellensburg
This paper presents implementation of Data Mining and Knowledge Discovery techniques for the search for regularities in tables of context features of DNA sequences involved in transcription regulation. The goal is to discover regularities, which connect nucleotide sequences with the functional class of those sequences. The search patterns for regularities have been constructed in first-order logic augmented by probabilistic estimates. The PC software system "Gene Discovery" has been designed for this purpose. This system accepts molecular-genetics data retrieved from the database using SQL queries. Sequences of erythroid-specific promoters and promoters of genes of endocrine system from TRRD database (Kolchanov N.A. et al., 2000) have been analysed by this system. Several regularities that connect (i) the nucleotide sequences in the regulatory DNA and its location relative to transcription start with (ii) functional class have been found. A method for recognition of the DNA promoter class has been developed using these regularities.
Ключевые слова: Машинное обучение (Machine Learning), Открытие Знаний (Knowledge Discovery), Анализ Данных (Data Mining), биоинформатика, распознавание промоторов эукариот, сайты связывания транскрипционных факторов
Исследование структуры промоторов представляет интерес для понимания механизмов транскрипции эукариот. Обязательным элементом, абсолютно необходимым для инициации транскрипции, является коровый (базальный) промотор, под которым понимают минимальную последовательность ДНК, необходимую для правильной инициации транскрипции гена in vitro. В коровый промотор входит старт транскрипции и область приблизительно от -60 до +40 п.о. по отношению к нему (Zhang M.Q., 1998; Arnone M.I. & Davidson E.H., 1997). Каждый регуляторный район содержит в своем составе сайты связывания определенных транскрипционных факторов (ССТФ) (Nikolov D.B. & Burley S.K., 1997). Один ген может иметь множество промоторов, определяющих формирование различных белковых продуктов или обладающих различным уровнем специфической функциональной активности. Кроме того, для промоторов эукариот характерно отсутствие точной локализации контекстных сигналов, значимых для их функционирования и слабость этих сигналов (Goodrich J.A. et al., 1996).
Разнообразие строения промоторных районов генов создаёт наибольшие трудности для разработки программ распознавания промоторов. Несмотря на то, что создано большое количество методов распознавания промоторов РНК полимеразы II в геномах эукариот (Pedersen A.G. et al., 1999; Kondrakhin Y.V. et al., 1995) проблема повышения точности распознавания в целом остается нерешенной. Существующие методы распознавания основаны на весовых матрицах или других мерах сходства анализируемой последовательности к обучающей выборке (базе данных известных последовательностей). В то же время выбор участка последовательности для оценки сходства основан на экспертных биологических знаниях. В то же время эти знания не были независимо проверены статистически. (Например, предположение о значимости участка от -40 до -20 п.о. относительно старта транскрипции значимо для промоторов, содержащих TATA-бокс, и в то же время не обязательно для TATA-несодержащих промоторов).
Анализ данных в других научных областях связан с экстенсивной обработкой больших баз данных и открытием больших наборов закономерностей. В то же время, информация, распределенная по научной литературе и сосредоточенная в молекулярно-биологических базах данных, содержит тысячи экспериментальных результатов о последовательностях ДНК, вовлеченных в регуляцию транскрипции. В настоящее время в мире существует около 300 молекулярно-биологических баз данных, доступных через Интернет. (Baxevanis A.D., 2001). Такое положение дает возможность широкомасштабного применения теории анализа данных и открытия знаний в биоинформатики (Jakobsen I.B. et al., 2001).
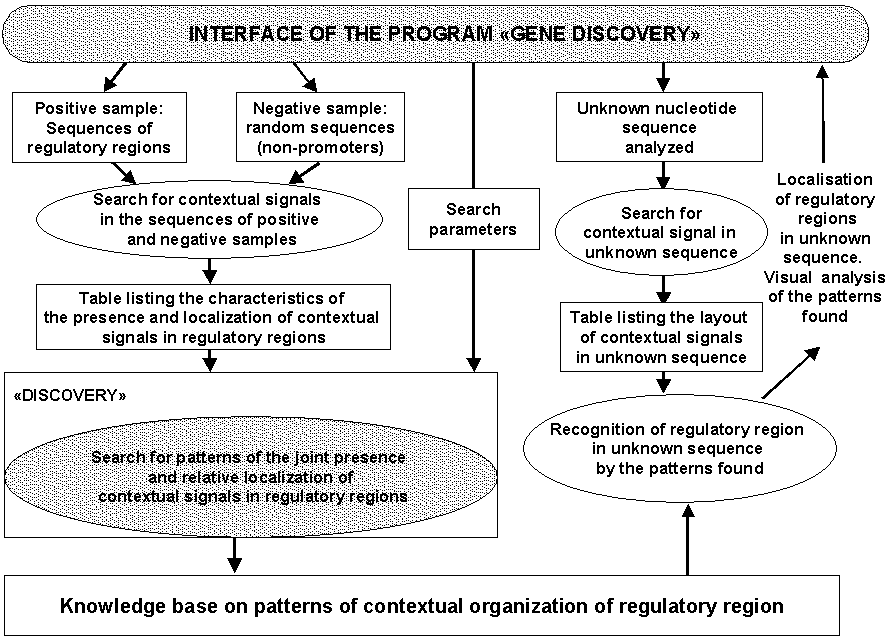
Компьютерная программа "Gene Discovery" была разработана для анализа структурной организации промоторов эукариот на основе информации об экспериментально известных и предсказанных функциональных сайтах.
Компьютерная система "Gene Discovery" является адаптацией системы "Discovery" для задач молекулярной биологии (Kovalerchuk B. & Vityaev E., 2000). "Gene Discovery" состоит из трех основных модулей: (1) модуль для интерактивного представления контекстных сигналов в стандартной таблице данных; (2) модуль "Discovery" для поиска закономерностей; (3) модуль для распознавания класса последовательности, используя найденные закономерности. Программа написана на языке С++ и предназначена для интерактивного использования. Блок анализа данных "Discovery" обозначен на рисунке 1 как "поиск паттернов совместного присутствия и взаимной локализации контекстных сигналов" ("Search for patterns of the joint presence and relative localisation of contextual signals...") Другие модули системы предназначены для подготовки и интерпретации молекулярно-генетических данных.
Метод машинного обучения и созданная на его основе система "Discovery" (Витяев Е.Е. & Москвитин А.А., 1993) находит статистически значимые правила в логике первого порядка для функциональной аннотации регуляторных районов. Система "Discovery" успешно применялась к решению многих проблем в психологии, физике, медицине, финансах и других науках (Kovalerchuk B. & Vityaev E. et al, 1997, 2000, 2001) (см. также www-сайт "Scientific Discovery": http://www.math.nsc.ru/LBRT/l/vityaev/, раздел "comparison"). Также как и любая техника, основанная на логических правилах, данная техника позволяет получить предсказывающие правила на естественном языке, которые интепретируются с биологической точки зрения и обеспечивают предсказание промоторов (функциональную аннотацию) (Mitchell T., 1997). Биолог может оценить корректность распознавания и значимость правил самих по себе. Научной проблемой в применении предсказывающих систем, основанных на данных, является обобщение. Система "Discovery" обобщает данные через обнаружение логических вероятностных правил-законов.
Концепция вероятностной обусловленности (P.Suppes, 1970) является
краеугольным камнем этого подхода при выводе из того, что ![]() есть
вероятностная причина
есть
вероятностная причина ![]() (
(
![]() ), когда вероятность
), когда вероятность ![]() при данном
при данном
![]() больше вероятности
больше вероятности ![]() . Система "Discovery" использует обобщенную версию
этой концепции. Она оперирует с логическими выражениями вида
. Система "Discovery" использует обобщенную версию
этой концепции. Она оперирует с логическими выражениями вида
![]() . Выражения
. Выражения
![]() позволяют заключать, что они являются
вероятностной причиной
позволяют заключать, что они являются
вероятностной причиной ![]() при выполнении некоторого
дополнительного условия, которое будет приведено ниже. Это условие требует,
чтобы условная вероятность
при выполнении некоторого
дополнительного условия, которое будет приведено ниже. Это условие требует,
чтобы условная вероятность
![]() события
события ![]() при любом подусловии
при любом подусловии
![]() была строго меньше, чем условная вероятность
была строго меньше, чем условная вероятность
![]() события
события ![]() при полном
условии. В частности, для выражения
при полном
условии. В частности, для выражения
![]() , если мы имеем
, если мы имеем
![]() , то условная вероятность
, то условная вероятность
![]() должна
быть строго меньше, чем
должна
быть строго меньше, чем
![]() -- вероятность
-- вероятность ![]() при данном
при данном ![]() .
Эта идея приводит нас к следующему определению (Kovalerchuk B. & Vityaev
E., 2000):
.
Эта идея приводит нас к следующему определению (Kovalerchuk B. & Vityaev
E., 2000):
Определение: правило
![]() является правилом-законом (выражает
вероятностную причинную зависимость между посылкой
является правилом-законом (выражает
вероятностную причинную зависимость между посылкой
![]() и заключением
и заключением ![]() , если:
, если:
![]() (не равно)
(не равно)
![]() .
.
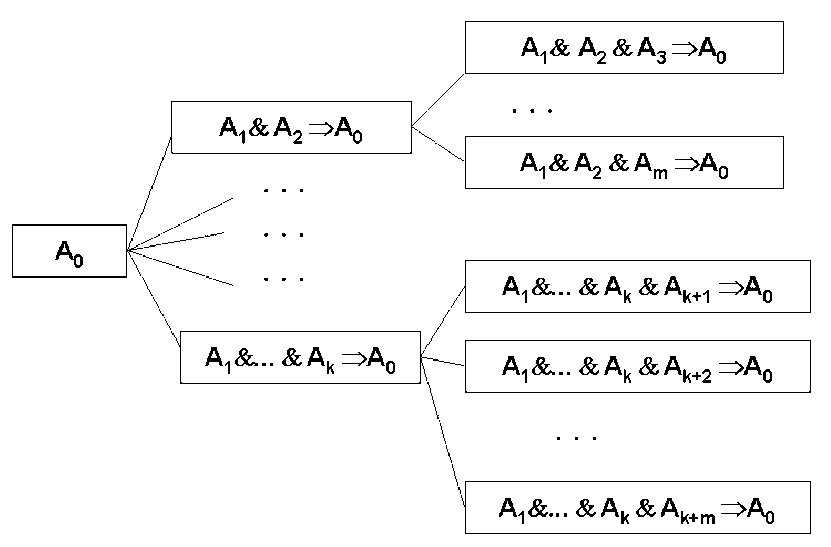
Это определение правила-закона удовлетворяет всем свойствам научных законов. Концептуально правила-законы пришли из философии науки. Эти правила пытаются зафиксировать математически существенные особенности научных законов: (1) высокий уровень обобщения; (2) простоту (Бритва Оккама); и (3) максимальная опровержимость.
Правило
![]() позволяет генерировать подправила
позволяет генерировать подправила
1) Подправило более общее (логически сильнее и описывает тот же набор событий);
2) Подправило проще, чем само правило, поскольку состоит из меньшего числа утверждений в условной части;
3) Подправило более опровержимо, чем само правило, поскольку больший набор возможных примеров может его опровергнуть (условная часть подправила имеет меньше ограничений).
Таким образом, если правило ![]() покрывает набор примеров, то нужно проверить,
что бы ни одно из его подправил
покрывает набор примеров, то нужно проверить,
что бы ни одно из его подправил ![]() не покрывали тот же набор примеров. В
противном случае это подправило (или, возможно, некоторое его подправило)
будет предпочтительнее, поскольку это подправило проще, более общо и более
опровержимо. В детерминистическом случае правило-закон может быть
определено (для некоторого набора примеров) как правило без подправил,
покрывающих тот же набор примеров. Другими словами, правило-закон есть
правило, которое истинно для некоторого набора примеров, но ни одно из его
подправил не истинно для этого же набора примеров.
не покрывали тот же набор примеров. В
противном случае это подправило (или, возможно, некоторое его подправило)
будет предпочтительнее, поскольку это подправило проще, более общо и более
опровержимо. В детерминистическом случае правило-закон может быть
определено (для некоторого набора примеров) как правило без подправил,
покрывающих тот же набор примеров. Другими словами, правило-закон есть
правило, которое истинно для некоторого набора примеров, но ни одно из его
подправил не истинно для этого же набора примеров.
Если примеры содержат шум, что типично для естественных наук, следует
использовать вероятностные характеристики выражений вместо значений
истина/ложь. Условная вероятность правила используется в системе
"Discovery" в качестве такой характеристики. Условная вероятность правила
![]() определена как
определена как
![]() , предполагая, что
, предполагая, что
![]() . Подобным же образом определены условные вероятности
. Подобным же образом определены условные вероятности
![]() для подправил
для подправил
![]() , предполагая, что
, предполагая, что
![]() . Условная вероятность Prob(C)
используется далее для оценки прогностической силы правила для предсказания
. Условная вероятность Prob(C)
используется далее для оценки прогностической силы правила для предсказания
![]() . Кроме того, условная вероятность - основное средство для
определения не детерминистических (вероятностных) правил-законов
(Kovalerchuk B. et al., 2001; Витяев Е.Е., 1992).
. Кроме того, условная вероятность - основное средство для
определения не детерминистических (вероятностных) правил-законов
(Kovalerchuk B. et al., 2001; Витяев Е.Е., 1992).
Правило является вероятностным правилом-законом тогда и только тогда, когда все его подправила имеют значимую более низкую условную вероятность, чем само правило. Другое определение правил-законов может быть получено в терминах обобщения. Правило является правилом-законом тогда и только тогда, когда оно не может быть обобщено без статистически значимого уменьшения его условной вероятности. Правила-законы определены таким образом, чтобы соответствовать всем трем отмеченным выше свойствам (свойства научных законов), т.е., эти правила (1) общие с логической точки зрения, (2) простые, и (3) опровержимые.
Система "Discovery" ищет все цепочки
![]() связанных подправил-законов,
где
связанных подправил-законов,
где ![]() есть подправило
есть подправило
![]() - подправило правила
- подправило правила
![]() и, окончательно,
и, окончательно,
![]() - подправило правила
- подправило правила
![]() . Также
. Также
![]() . Доказана теорема
(Витяев Е.Е., 1992) о том, что все правила, имеющие максимальные значения
условной вероятности, могут быть найдены в конце таких цепочек.
. Доказана теорема
(Витяев Е.Е., 1992) о том, что все правила, имеющие максимальные значения
условной вероятности, могут быть найдены в конце таких цепочек.
Цель системы "Discovery" -- найти все наиболее сильные предсказывающие правила (St-правила (strong)).
Определение 1. Правило
![]() , где
, где ![]() и
и
![]() называется St-правилом для атомарной формулы
называется St-правилом для атомарной формулы ![]() на данных
на данных ![]() , если и
только если:
, если и
только если:
1)
![]() , где
, где
![]() - условия, выполненные
на данных
- условия, выполненные
на данных ![]() ;
;
2) Правило ![]() имеет максимум условной вероятности
имеет максимум условной вероятности ![]() среди правил
среди правил ![]() ,
удовлетворяющих условию 1 и выполненных на тех же данных, что и
,
удовлетворяющих условию 1 и выполненных на тех же данных, что и ![]() , и
, и
3) Для любого правила ![]() , удовлетворяющего условиям 1, 2, мы имеем
, удовлетворяющего условиям 1, 2, мы имеем
![]() .
.
Условие 1 означает, что St-правило ![]() имеет значимую условную часть (посылку),
т.е.
условная вероятность
имеет значимую условную часть (посылку),
т.е.
условная вероятность ![]() правила
правила ![]() больше, чем вероятность
атомарной формулы
больше, чем вероятность
атомарной формулы ![]() как таковой. Если это условие не выполняется, то нет
оснований добавлять посылку для прогнозирования
как таковой. Если это условие не выполняется, то нет
оснований добавлять посылку для прогнозирования ![]() . Если атом A сам имеет
высокую вероятность, то он может быть предсказан без посылки
. Если атом A сам имеет
высокую вероятность, то он может быть предсказан без посылки
Условие 2 приводит к "сильнейшему" St-правилу, т.е. правилу с максимумом условной вероятности среди правил, удовлетворяющих указанному выше условию 1 на тех же данных.
Условие 3 означает, что St-правило С является наиболее "общим" среди
правил, удовлетворяющих условиям 1 и 2, т.е., правило ![]() охватывает наиболее
широкий набор случаев из
охватывает наиболее
широкий набор случаев из ![]() , для которых оно применимо.
, для которых оно применимо.
Доказана теорема (Витяев Е.Е., 1992) что все St-правила
могут быть найдены в конце цепочек
![]() . Так достигается цель системы
"Discovery".
. Так достигается цель системы
"Discovery".
Алгоритм перестает генерировать новые правила, когда они становятся слишком сложными (т.е. статистически незначимыми для анализируемых данных) даже если правила имеют высокую точность на обучении. Для оценки статистической значимости в алгоритме используется статистический критерий Фишера (точный критерий Фишера для таблиц сопряженности) (Kendall M.G. & Stuart A., 1977).
Другой очевидный критерий остановки - ограничение числа условий ![]() (число полей данных в анализируемой таблице).
(число полей данных в анализируемой таблице).
Теоретические преимущества обобщения представлены в теореме (Витяев Е.Е., 1992; Витяев Е.Е. & Москвитин А.А., 1993). Данный подход имеет сходство с подходом подсказок (Abu-Mostafa, 1990). Мы используем математический формализм правил логики первого порядка, описанный в ряде работ (Russel S. & Norvig P., 1995; Halpern J.Y., 1990; Krantz D.H. et al. 1971, 1989, 1990). Заметим, что класс правил логики первого порядка шире, чем класс решающих деревьев. (Mitchell T., 1997).
Компьютерная система "Gene Discovery" является адаптацией системы "Discovery" для анализа нуклеотидных последовательностей регуляторных районов. Принципиальная схема системы представлена на рисунке 1.
На вход системы подается обучающая выборка нуклеотидных последовательностей двух альтернативных классов: класс 1 - промоторы; класс 2 - последовательности, не выполняющие этой функции (например, случайные последовательности с теми же частотами нуклеотидов, либо соседние участки последовательности, не несущие регуляторной функции, например, экзоны).
Имеется блок программ, осуществляющих поиск контекстных сигналов в последовательностях этих двух классов (рисунок 1). Сигнал может быть:
контекстным (короткое олигонуклеотидное слово, функциональный сайт и т.д.),
конформационным (участок ДНК, характеризующийся особенностями конформационных или физико-химических свойств, например, легкоплавкие участки ДНК, сильно изогнутая ДНК и т.д.),
структурным (например, Z-ДНК или шпилька вторичной структуры РНК и др.).
Все эти сигналы могут быть установлены с использованием знаний о свойствах ДНК и консенсусных схемах, на основе экспериментальной информации из специализированных баз данных.
В настоящей работе рассматриваются контекстные сигналы только одного типа - несовершенные олигонуклеотиды.
С помощью этой программы были проанализированы последовательности промоторов генов эндокринной системы и соответствующие им по частотам олигонуклеотидов случайные последовательности. Выборка 40 промоторов была взята из базы данных ES-TRRD (http://wwwmgs.bionet.nsc.ru/). Последовательности промоторов имели длину 120 п.о. (от -100 до +20 п.о. относительно старта транскрипции). Оценка гомологии между промоторами показала, что она для любой пары не превышает 60% . Негативная выборка "не-промоторов" содержала 1000 случайных последовательностей той же длины и с теми же частотами нуклеотидов, что и последовательности промоторов. Для генерации случайных последовательностей использовалась программа http://wwwmgs.bionet.nsc.ru/mgs/dbases/nsamples/.
Для выделения олигонуклеотидных сигналов, специфичных к данной группе промоторов, использовалась программа ARGO (Babenko V.N. et al., 1999), (см. также http://wwwmgs.bionet.nsc.ru/mgs/programs/argo/). Под олигонуклеотидным сигналом, или мотивом, понимается слово длиной 8 оснований, записанное в обобщённом 15-буквенном алфавите IUPAC: {A, T, G, C, R=G/A, Y=T/C, M=A/C, K=T/G, W=A/T, S=G/C, B=T/C/G, V=A/G/C, H=A/T/C, D=A/T/G, N=A/T/G/C}. Это стандартный способ представления близких строк нуклеотидов одной записью, как один сигнал. Таким образом, был выполнен подготовительный этап подготовки данных. Заметим, что подобные сигналы могут быть построены по гомологии с известными белковыми сайтами связывания в других формах записи.
Отобранные контекстные сигналы (вырожденные олигонуклеотиды) были локазизованы в исследуемых последовательностях ДНК и представлены в виде таблицы данных с помощью модуля "Gene Discovery". Итак, данные были представлены в виде таблицы "объект-признак". В этой таблице объектами являются последовательности ДНК, признаками - присутствие контекстных сигналов и их локализация относительно экспериментально определенного старта транскрипции.
Таким же образом была проанализирована другая выборка промоторов, а именно,
промоторы эритроидных генов. В итоге для анализа данных была построена
таблица, содержащая несколько тысяч строк. Она содержала последовательность
контекстного сигнала ![]() и его позицию
и его позицию
![]() в
промоторном районе. Например, для первого промотора в анализируемой выборке
сигнал
в
промоторном районе. Например, для первого промотора в анализируемой выборке
сигнал ![]() =TGACCAAT,
=TGACCAAT,
![]() , сигнал
, сигнал ![]() =RCCAATND,
=RCCAATND,
![]() и т.д.
и т.д.
Проверяемая гипотеза ![]() в нашем случае звучит так:
"Принадлежит ли последовательность к классу 1 (промоторы)?"
в нашем случае звучит так:
"Принадлежит ли последовательность к классу 1 (промоторы)?"
Назовем комплексным сигналом группу олигонуклеотидных мотивов, имеющих
некоторую схему взаимного расположения в промоторной последовательности.
Другими словами комплексный сигнал - это группа описанных ранее коротких
контекстных сигналов. Присутствие такого комплексного сигнала может
рассматриваться как условие ![]() принадлежности к классу
промоторов. Рассмотрим простейший комплексный сигнал
принадлежности к классу
промоторов. Рассмотрим простейший комплексный сигнал
![]() , образованный парой олигонуклеотидов с заданным порядком:
, образованный парой олигонуклеотидов с заданным порядком:
То есть мы можем рассматривать условие ![]() как
как
![]() , и проверить гипотезу
, и проверить гипотезу
![]() для всех последовательностей ДНК, содержащих
для всех последовательностей ДНК, содержащих ![]() и
и ![]() .
.
Но присутствие только двух олигонуклеотидов (
![]() может быть недостаточным условием. Поэтому мы должны рассмотреть все тройки
олигонуклеотидов, такие как
может быть недостаточным условием. Поэтому мы должны рассмотреть все тройки
олигонуклеотидов, такие как
![]() . Формально такая тройка может быть рассмотрена как две пары
. Формально такая тройка может быть рассмотрена как две пары
![]() и
и
![]() . Гипотеза
для тестирования сейчас
. Гипотеза
для тестирования сейчас
![]() . Таким образом, используя логику первого порядка, мы строим более и
более сложные условия, включая присутствие этих олигонуклеотидов в прямой
или комплементарной цепи ДНК, перекрывание олигонуклеотидов (пересечение
позиций) и так далее.
. Таким образом, используя логику первого порядка, мы строим более и
более сложные условия, включая присутствие этих олигонуклеотидов в прямой
или комплементарной цепи ДНК, перекрывание олигонуклеотидов (пересечение
позиций) и так далее.
В результате анализа "Gene Discovery" было найдено большое число закономерностей совместного появления контекстных сигналов в промоторных районах. Число закономерностей зависит от параметров поиска, задаваемых пользователем. Если мы определим низкий уровень условной вероятности (менее 0.5), то итоговое число правил будет очень большим (до нескольких тысяч). Такое число правил трудно интерпретировать эксперту. Поэтому мы можем задать высокий уровень условной вероятности, например, более 0.95. Тогда число правил будет мало, но они будут очень значимы с биологической точки зрения.
Найденные закономерности могут быть проанализированы экспертом в молекулярной биологии как уникальные комплексные сигналы, значимые для правильного функционирования промотора.
| 1 | 2 | 3 | 4 | 5 | |
| 1 | CWGNRGCN<NGSYMTAM<CAGGRNCH | 0.875 | 0.00054 | 4 | 0.24 (<1) |
| 2 | KGRSSAGR<CYCYNSCY<CWGSNYCH | 1.0 | 0.00012 | 4 | 0.28 (<1) |
| 3 | CWGNRGCN<NGSYMTAM<MAGKSHCN | 1.0 | 0.00009 | 6 | 0.47 (<1) |
| 4 | CWGNRGCN<NGSYMTAM<CMDGGNCH | 0.846 | 0.00099 | 5 | 0.43 (<1) |
| 5 | CNKSAGNT<NCARGRNC<HNNKGCTG | 1.0 | 0.01426 | 4 | 0.37 (<1) |
| 6 | RNWGGCCN<DGRGNRGG<TCMAGNMN | 0.875 | 0.00118 | 4 | 0.4 (<1) |
| 7 | RGSNRGRG<NNGSTWTA<CNCNRKGC | 1.0 | 0.02852 | 5 | 0.53 (<1) |
| 8 | NNGSTWTA<NMAGDGMC<CNCNRKGC | 0.875 | 0.04755 | 5 | 0.53 (<1) |
| 9 | RGSNRGRG<NNGSTWTA<CMDGGNCH | 1.0 | 0.03964 | 5 | 0.55 (<1) |
| 10 | RGSNRGRG<KGGNSAGD<ANCTSMNG | 1.0 | 0.03964 | 4 | 0.45 (<1) |
| ... | ... | ... | ... | ... | ... |
| 45 | RGSNRGRG<NGSYMTAM<CNCNRKGC | 1.0 | 0.03964 | 5 | 0.58 (<1) |
Примечания. Данные в таблице приведены не полностью из-за большого объема, пропуски обозначены многоточиями.
1 - Комплексный сигнал состоит из олигонуклеотидов в 15-буквенном алфавите, линейно расположенных на последовательности в соответствии с приведенной записью. Знак "<" означает, что позиция первого олигонуклеотида относительно старта транскрипции меньше позиции второго. Расстояние между отдельными сигналами не фиксировано.
2 - Условная вероятность
![]() считается как
отношение числа промоторов, имеющих данный сигнал
считается как
отношение числа промоторов, имеющих данный сигнал ![]() к общему
числу последовательностей, имеющих данный сигнал
к общему
числу последовательностей, имеющих данный сигнал
![]() .
.
3 - Оценка вероятности получить сигнал в промоторах по случайным причинам
большее число раз, чем наблюдаемое, по точному критерию Фишера для таблиц
сопряженности
![]() .
.
4 - Количество промоторов в обучающей выборке, имеющих данный комплексный сигнал.
5 - Ожидаемое по случайным причинам количество промоторов, имеющих комплексный сигнал. В предположении независимости входящих в комплексный сигнал олигонуклеотидов оценивается как произведение общего числа промоторов на частоты олигонуклеотидов в промоторах, с учетом вариантов их взаимного линейного расположения.
Рассмотрим дополнительные условия к отбору комплексных сигналов, помимо высокого уровня условной вероятности:
(1) индивидуальные сигналы, входящие в комплексный сигнал, не пересекаются на последовательностях рассмотренных промоторов;
(2) наблюдаемое количество промоторов ![]() , в которых встретился комплексный
сигнал выше числа
, в которых встретился комплексный
сигнал выше числа ![]() , которое по случайным причинам ожидается в выборке
промоторов,
, которое по случайным причинам ожидается в выборке
промоторов, ![]() .
.
Ожидаемое количество ![]() оценивалось как произведение частот отдельных
олигонуклеотидов в промоторах, умноженное на общее число промоторов, с
учетом числа вариантов взаимного расположения олигонуклеотидов на
последовательности промотора. Например, ожидаемое количество промоторов
оценивалось как произведение частот отдельных
олигонуклеотидов в промоторах, умноженное на общее число промоторов, с
учетом числа вариантов взаимного расположения олигонуклеотидов на
последовательности промотора. Например, ожидаемое количество промоторов ![]() ,
в которых встретился комплексный сигнал
,
в которых встретился комплексный сигнал
![]() , равно
, равно
Примеры комплексных сигналов, удовлетворяющих этим условиям, специфичных для
промоторов генов эндокринной системы приведены в таблице 1. Рассмотрим
сигнал CWGNRGCN<NGSYMTAM<MAGKSHCN. Знак "<" здесь означает, что позиции
соответствующих олигонуклеотидов упорядочены относительно старта
транскрипции. Итак, ожидаемое число встретить этот сигнал ![]() , т.е.
меньше единицы, в то время как он встретился в 6 промоторах, что в
приблизительно в 13 раз больше ожидаемого уровня.
, т.е.
меньше единицы, в то время как он встретился в 6 промоторах, что в
приблизительно в 13 раз больше ожидаемого уровня.
Пример расположения комплексного сигнала представлен на рисунке 3.
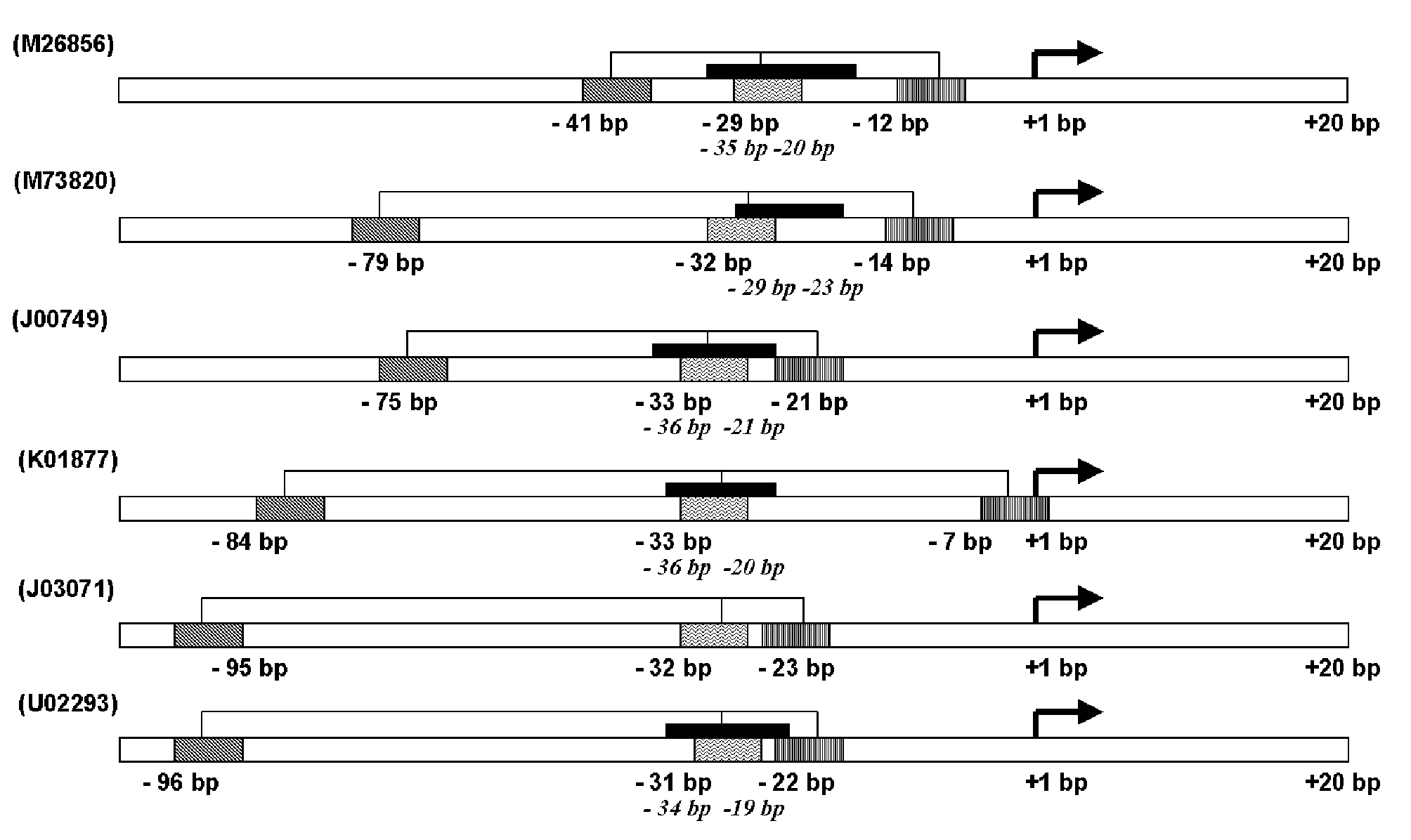 |
Промоторные последовательности выровнены относительно старта транскрипции (позиция +1 п.о.), отмеченная стрелкой. Идентификаторы EMBL рассмотренных последовательностей приведены в скобках. Восьминуклеотидные мотивы, представляющие комплексные сигналы показаны как затемненные прямоугольники. Указана позиция первого олигонуклеотида относительно старта транскрипции. Черным прямоугольниками показаны позиции TATA-бокса, проиндексированные в базе данных TRRD; позиции первого и последнего нуклеотида выделены курсивом.
Следует заметить, что система не переобучается на обучающих примерах и показывает ошибку первого рода на контроле примерно равную условной вероятности правил, вычисленных на обучении.
Таким образом, разработанная нами компьютерная система "Gene Discovery" позволяет выявлять как индивидуальные значимые мотивы (вырожденные квазиинвариантные олигонуклеотиды), так и комплексные сигналы. Проведенный анализ показал, что промоторы генов эндокринной системы и эритроид-специфичные промоторы характеризуются высокой насыщенностью такими сигналами. О функциональной значимости комплексных сигналов свидетельствует тот факт, что они имеют сходное расположение в пределах подгрупп специфичных промоторов. Кроме того, как отмечалось выше, комплексные сигналы могут иметь сходные расстояния между индивидуальными мотивами. При этом анализируемые промоторы не имеют выраженной гомологии.
Индивидуальные мотивы могут соответствовать сайтам связывания транскрипционных факторов. Еще в ранних работах по анализу и распознаванию промоторов было показано, что они обогащены потенциальными сайтами связывания транскрипционных факторов по сравнению со случайными последовательностями (Kondrakhin Yu.V. et al., 1995). Индивидуальные мотивы могут также соответствовать участкам ДНК, обеспечивающим специфические конформационные или физико-химические свойства: повышенную гибкость ДНК, легкоплавкость и т.д., необходимые для функционирования промоторов.
При рассмотрении комплексных сигналов следует отметить несколько обстоятельств.
Во-первых, в ряде работ (см. (Kondrakhin Yu.V. et al., 1995; Klingenhoff A. et al., 1999) выявлены специфичные паттерны распределения потенциальных сайтов связывания транскрипционных факторов с максимумами локализации различных сайтов в различных участках промоторов. Таким образом, наблюдающиеся комплексные сигналы могут отражать преимущественное расположение различных сайтов в определенных участках промоторов. Доказательства важности позиционирования контекстных характеристик в пределах промоторов также получены М.Зангом (Zhang M.Q., 1998). В работах В.Г.Левицкого (Левицкий В.Г. и др., 2000) выявлено разбиение промотора на локальные участки с характерным динуклеотидным составом.
Во-вторых, в последнее время активно изучается особый тип регуляторных элементов, контролирующих транскрипцию, которые называются композиционными элементами (КЭ)(Kel-Margoulis O.V. et al., 2000). Они образованы парами сайтов связывания транскрипционных факторов, которые в результате белок-белковых взаимодействий между соответствующими транскрипционными факторами приобретают новые регуляторные свойства. Каждый из сайтов в составе КЭ способен функционировать по отдельности, но их взаимодействие обеспечивает существенно более выраженный активирующий или репрессирующий эффект на транскрипцию гена. В настоящее время экспериментально выявлено более 150 КЭ (Kel-Margoulis O.V. et al., 2000) (см. также http://compel.bionet.nsc.ru/). Исследование закономерностей совместной встречаемости и взаимного расположения сайтов c помощью системы "Gene Discovery" открывает путь для создания компьютерных методов поиска потенциальных композиционных элементов. Мы полагаем, что выявление и учет комплексных сигналов позволит в дальнейшем существенно повысить точность распознавания специфических групп промоторов.
Авторы выражают признательность В.Г. Левицкому, Е.В. Игнатьевой, О.А. Подколодной за ценные замечания и помощь в работе.
 Ваши комментарии
Ваши комментарии
|
![[SBRAS]](/gif/s_sigma.gif)
[Головная страница] [Конференции] [СО РАН] |
© 2001, Сибирское отделение Российской академии наук, Новосибирск
© 2001, Объединенный институт информатики СО РАН, Новосибирск
© 2001, Институт вычислительных технологий СО РАН, Новосибирск
© 2001, Институт систем информатики СО РАН, Новосибирск
© 2001, Институт математики СО РАН, Новосибирск
© 2001, Институт цитологии и генетики СО РАН, Новосибирск
© 2001, Институт вычислительной математики и математической геофизики СО РАН, Новосибирск
© 2001, Новосибирский государственный университет
Дата последней модификации Friday, 14-Sep-2001 16:23:09 NOVST